
Скрининг ребенка 2 года

[13-070]
Целиакия. Скрининг (дети до 2 лет)
3550 руб.
Определение уровня антител IgA к тканевой трансглутаминазе и антител IgA и IgG к дезаминированным пептидам глиадина, используемое для уточнения диагноза “целиакия” (“глютеновая энтеропатия”).
Состав исследования:
- 13-034 Антитела к тканевой трансглутаминазе, IgA
- 13-133 Антитела к дезаминированным пептидам глиадина, IgA
- 13-134 Антитела к дезаминированным пептидам глиадина, IgG
Синонимы русские
Уточнение диагноза “глютеновая энтеропатия”.
Синонимы английские
Celiac disease (gluten-sensitive enteropathy), definitive serologic tests.
Метод исследования
Непрямая реакция иммунофлюоресценции.
Какой биоматериал можно использовать для исследования?
Венозную кровь.
Как правильно подготовиться к исследованию?
- Рекомендуется не придерживаться безглютеновой диеты в течение 7 дней до исследования.
- Не курить в течение 30 минут до исследования.
Общая информация об исследовании
Целиакия (глютеновая энтеропатия) – это аутоиммунное заболевание, возникающее у предрасположенных лиц в ответ на поступление с пищей глютена. Он присутствует во многих продуктах питания, богатых злаковыми (ячмень, рожь и пшеница). Наибольшее значение в развитии аутоиммунного процесса имеет один из компонентов глютена – белок глиадин. Под воздействием фермента тканевой трансглутаминазы глиадин превращается в более иммуногенный пептид, а также образует иммуногенные комплексы с этим ферментом. Поступление с пищей глютена провоцирует и поддерживает воспалительный процесс в слизистой оболочке кишки, который с течением времени приводит к ее атрофии. Наблюдаемые при этом морфологические и клинические признаки объединяют в понятие глютеновой энтеропатии.
При этом заболевании вырабатываются разнообразные антитела, в том числе к глиадину, эндомизию, тканевой трансглутаминазе и другим компонентам. Определение уровня этих антител используется для диагностики целиакии.
Для скрининга целиакии используют комбинацию следующих серологических тестов: антитела к тканевой трансглутаминазе IgA и антитела к дезаминированным пептидам глиадина IgA и IgG.
Антитела к тканевой трансглутаминазе IgA и IgG направлены против фермента, участвующего в биохимических превращениях глиадина. Специфичность определения IgA-антител к тканевой трансглутаминазе в отношении целиакии выше, чем специфичность теста на IgG (97-99 % против 77-95 %).
Антитела к дезаминированным пептидам глиадина относятся к лабораторным серологическим показателям, выявление которых используется в диагностике глютен-чувствительной энтеропатии (целиакии). Дезаминированные пептиды глиадина являются мощными активаторами иммунной системы человека, в результате чего возникает иммунный ответ и формируются аутоантитела. Антитела к дезаминированным пептидам глиадина представлены иммуноглобулинами классов A и G (IgA, IgG). Данные антитела используются в комплексной в диагностике целиакии наряду с выявлением антител к тканевой трансглутаминазе, IgA/IgG и антител к эндомизию. Подтверждение диагноза “целиакия” возможно только после проведения морфологического исследования, биопсии тонкой кишки и выявления определенных изменений исследуемого биоматериала. Тесты по выявлению антител к дезаминированным пептидам глиадина класса IgA обладают высокими параметрами чувствительности и специфичности в сравнении с тестами по выявлению антител к альфа-глиадину. Диагностическая чувствительность составляет 80-90 %, диагностическая специфичность 95-100 %. Антитела к дезаминированным пептидам глиадина класса IgA рекомендуется определять в комбинации с антителами класса IgG до назначения безглютеновой диеты. Их выявление используется в качестве дополнительных параметров в комплексной диагностике целиакии. Выявление антител класса IgA имеет большее диагностическое значение у детей до двух лет, чем у взрослых лиц.
Уточнение диагноза “целиакия” проводится при наличии у пациента клинических признаков этого заболевания. В некоторых случаях оно также может потребоваться при отрицательном результате скрининга на целиакию.
Основная цель уточнения диагноза “целиакия” – выявить пациентов, подлежащих в дальнейшем эндоскопическому исследованию и биопсии тонкой кишки. Так как эндоскопия – это инвазивное исследование, сопряженное с определенными рисками, в последнее время некоторыми клиницистами предлагается заменить этот этап диагностики всесторонним и полноценным лабораторным обследованием пациента.
Целиакия часто сочетается с другими аутоиммунными заболеваниями, такими как сахарный диабет 1-го типа, аутоиммунный тиреоидит Хашимото и системные заболевания соединительной ткани, и при положительном результате исследования и подтверждении диагноза “целиакия” проводят дополнительные лабораторные тесты, исключающие сопутствующие заболевания.
Целиакия характеризуется выраженной генетической предрасположенностью. Показано, что распространенность целиакии среди родственников пациента с этим заболеванием гораздо выше, чем в среднем в популяции. Поэтому при подтверждении диагноза “целиакия” целесообразно провести лабораторное обследование членов семьи больного.
Для чего используется исследование?
- Для уточнения диагноза “целиакия” у пациентов с положительным результатом скрининга на целиакию;
- для диагностики целиакии у пациентов с клиническими признаками этого заболевания;
- для диагностики целиакии у пациентов с отрицательным результатом скрининга целиакии.
Когда назначается исследование?
- При положительном результате скрининга целиакии;
- при подготовке к эндоскопическому исследованию;
- при наличии симптомов целиакии (рвота, диарея, задержка роста (у детей), боли в области живота, анемия, эпилепсия, атопический дерматит, ангулярный хейлит, афт, герпетиформный дерматит Дюринга, признаки дефицита витаминов.
Что означают результаты?
Референсные значения
1. Антитела к тканевой трансглутаминазе IgA
| Компонент | Референсные значения |
| Концентрация | |
| Интерпретация | не обнаружены |
2. Антитела к дезаминированным пептидам глиадина, IgA
| Компонент | Референсные значения |
| Концентрация | |
| Интерпретация | не обнаружены |
3. Антитела к дезаминированным пептидам глиадина, IgG
| Компонент | Референсные значения |
| Концентрация | |
| Интерпретация | не обнаружены |
Положительный результат:
- целиакия (глютеновая энтеропатия).
Отрицательный результат:
- отсутствие глютеновой энтеропатии.
Что может влиять на результат?
- Безглютеновая диета до исследования на анализ может приводить к ложноотрицательному результату.
- У детей с целиакией исследование антител к тканевой трансглутаминазе IgA характеризуется более частыми ложноотрицательными результатами (особенно в возрасте до 3 лет).
- Уровень IgA-антител к тканевой трансглутаминазе может колебаться у одного и того же пациента, поэтому в некоторых случаях целесообразно проведение повторных анализов.
- Результат анализа на IgA-антитела к тканевой трансглутаминазе у пациентов с сочетанием целиакии и изолированного дефицита иммуноглобулина А всегда ложноотрицательный.
Важные замечания
- Результат анализа следует оценивать вместе с данными дополнительных анамнестических, лабораторных и инструментальных исследований.
Также рекомендуется
- Антитела к глиадину, IgA
- Антитела к глиадину, IgG
- Суммарные иммуноглобулины A (IgA) в сыворотке
- Целиакия. Расширенное серологическое обследование
- Копрограмма
Кто назначает исследование?
Гастроэнтеролог, врач общей практики, педиатр, дерматовенеролог.
Литература
- Armstrong D, Don-Wauchope AC, Verdu EF. Testing for gluten-related disorders in clinical practice: the role of serology in managing thespectrum of gluten sensitivity. Can J Gastroenterol. 2011 Apr;25(4):193-7.
- Dahlbom I, Olsson M, Forooz NK, Sjöholm AG, Truedsson L, Hansson T. Immunoglobulin G (IgG) anti-tissue transglutaminase antibodies used as markers for IgA-deficient celiac disease patients. Clin Diagn Lab Immunol. 2005 Feb;12(2):254-8.
- Dahele AV, Aldhous MC, Humphreys K, Ghosh S. Serum IgA tissue transglutaminase antibodies in coeliac disease and other gastrointestinal diseases. QJM. 2001 Apr;94(4):195-205.
Источник
Неонатальный скрининг включен согласно рекомендациям ВОЗ в перечень обязательных медицинских тестов для малышей.
Подробнее об услуге…
Неонатальный скрининг обычно проводится в первые 10 дней жизни ребенка, но точная дата может существенно меняться в зависимости от обстоятельств.
Узнать цену услуги…
Скрининг наследственно обусловленных заболеваний обмена — медицинская услуга, которой можно воспользоваться в том числе без направления педиатра.
Где можно сделать скрининг?
Контроль качества лабораторных исследований, осуществляемый по международным стандартам, повышает вероятность получения точных и достоверных результатов анализов.
Что стоит учесть при выборе лаборатории?
Существенно сэкономить на медицинских диагностических услугах помогут акции и специальные предложения клиник.
Акции и скидки…
Главный вопрос, который волнует родителей новорожденного малыша, — здоровье ребенка. Многие врожденные болезни коварны и никак себя не проявляют на первом году жизни. Именно по этой причине был разработан метод исследования, известный как скрининг новорожденных. Это несложный тест, позволяющий выявить наличие многих врожденных заболеваний в первые дни жизни ребенка.
Что такое скрининг новорожденных
Скрининг новорожденных — это анализ крови, позволяющий провести раннюю диагностику как минимум 50 врожденных заболеваний. Метод является самым точным на сегодняшний день способом ранней диагностики генетически обусловленных патологий. По рекомендации ВОЗ скрининг новорожденных включен в перечень обязательных медицинских тестов для малышей.
На заметку
В России многие мамы называют скрининг новорожденных «пяточным тестом», поскольку кровь для него берут из пяточки малыша.
Следует знать, что положительный результат далеко не всегда означает, что у ребенка действительно есть то или иное заболевание. Для полной уверенности родителям выдадут направление на дополнительное обследование у соответствующего специалиста.
Когда проводится неонатальный скрининг у новорожденных
Это исследование проводится в первые 10 дней жизни ребенка, но точная дата может существенно меняться в зависимости от обстоятельств.
Скрининг новорожденных не рекомендуется делать в первые 2–3 дня жизни, поскольку при исследовании, проведенном так рано, есть риск ошибки — результат может быть ложноотрицательным или ложноположительным.
Обычно кровь на анализ у малышей, родившихся в срок, берут в роддоме на 4-ые сутки жизни. Недоношенным детям тест проводят на 7-ой день. Если мама с младенцем к этому времени уже выписалась из родильного отделения, кровь для скрининга берут на дому или в поликлинике.
Такие ранние сроки проведения анализа объясняются тем, что некоторые генетически обусловленные болезни проявляются в первые же недели жизни и крайне важно вовремя диагностировать их, чтобы начать терапию. Однако расширенный тест, включающий диагностику большего количества заболеваний, можно проводить и позже. Начиная с трехмесячного возраста кровь для анализа берут уже не из пятки, а из пальца.
Какие исследования входят в обязательную программу скрининга на наследственные заболевания
Поскольку болезней, которые могут быть выявлены посредством этого исследования, очень много, список патологий при обязательном скрининге сокращен до пяти самых распространенных:
- Гипотиреоз. Патология щитовидной железы, которая может привести к отставанию в физическом и психическом развитии. На сегодняшний день своевременно диагностированный гипотиреоз хорошо поддается гормональной терапии. Распространенность заболевания — 1 случай из 5 тысяч.
- Андрогенитальный синдром. Патология коры надпочечников, при которой нарушается нормальная выработка гормона кортизола. Может проявиться в виде задержки развития половой системы, проблем с сосудами и сердцем. Полному излечению этот синдром не поддается, но его можно держать под контролем при помощи гормональной терапии. Распространенность заболевания — 1 случай из 15 тысяч.
- Муковисцидоз. Заболевание проявляется заметным сгущением секрета в пищеварительном тракте и легких, что приводит к поражениям печени, ЖКТ, дыхательной системы и других органов. Поддается лечению. Распространенность заболевания — 1 случай из 3 тысяч.
- Фенилкетонурия. Заболевание, которое характеризуется нарушением выработки определенных ферментов. Последствия достаточно тяжелые. В первую очередь к ним относятся поражения ЦНС. Однако их можно избежать при помощи специальной диеты. Распространенность заболевания — 1 случай из 15 тысяч.
- Галактоземия. Так называют недостаток фермента, расщепляющего галактозу — один из сахаров, который содержится в лактозе и иных веществах. Последствия нехватки этого фермента проявляются через несколько недель жизни. У ребенка начинается желтуха, рвота, потеря аппетита. Со временем развиваются тяжелые патологии печени, замедляется умственное и физическое развитие, ухудшается зрение. Эта врожденная патология опасна, при этом встречается достаточно редко. Распространенность — 1 случай из 30 тысяч.
Данные заболевания были выбраны еще и потому, что при ранней диагностике их возможно вылечить или, по крайней мере, значительно облегчить последствия. Однако список патологий, которые можно определить при скрининге, намного шире, и при желании родители могут провести дополнительные тесты. В некоторых странах скрининг новорожденных включает диагностику большего количества заболеваний, например, в США — 40, а в Германии — 14.
Как подготовить малыша к исследованию
Чтобы результаты были максимально точными, кровь следует сдавать натощак, через 3 часа после последнего кормления. Анализ обычно проводят как минимум спустя 4 дня после начала грудного вскармливания. Подготовка к скринингу новорожденных очень проста. Перед забором крови ножку ребенка моют с мылом, протирают спиртом и насухо вытирают стерильной салфеткой — этим подготовительные меры и ограничиваются.
Как проводится забор крови
Эта процедура представляет собой довольно простой комплекс действий. На пяточке ребенка делается маленький прокол. Первая капля крови снимается стерильной салфеткой. Затем медсестра слегка сдавливает пяточку малыша и наносит полученную кровь на специальный бумажный тестовый бланк так, чтобы кровь пропитала пористую бумагу насквозь. В него вносится вся информация о новорожденном, а также об учреждении, где проводился забор крови. Бланк высушивается при комнатной температуре в течение 2–4 часов, помещается в конверт и отправляется в лабораторию или медико-генетический центр.
Интерпретация результатов анализа
На обработку образцов уходит 10–14 дней, после чего родители получают заключение генетической экспертизы. Результаты может интерпретировать только специалист. Помните, что результаты скрининга новорожденных — это еще не диагноз. Заключение может дать только врач соответствующего профиля на основании дополнительных исследований.
Несмотря на то, что скрининг новорожденных довольно точен, иногда он дает ложноположительные или ложноотрицательные результаты (чаще всего это связано с нарушением техники забора крови). Если результат анализа на какую-то болезнь положительный, родителям предлагают провести повторный тест. В случае повторного положительного результата малыш направляется на детальное обследование.
Полезная информация
Чаще всего ложноположительный результат при скрининге новорожденных дает тест на муковисцидоз.
Что такое расширенный неонатальный генетический скрининг методом ТМС?
Несмотря на то, что в программу обязательного скрининга новорожденных входит лишь пять заболеваний, генетически обусловленных болезней гораздо больше — около 500. К счастью, большинство врожденных патологий — большая редкость. Однако многие сознательные родители хотят получить как можно более полную информацию о здоровье ребенка и проходят расширенный неонатальный скрининг. Он позволяет выявить врожденные нарушения метаболизма в первые же недели жизни малыша.
Такой скрининг проводится методом тандемной масс-спектрометрии (ТМС) и дает возможность протестировать ребенка на 37 генетических заболеваний, среди которых — лейциноз, метилмалоновая ацидемия, недостаточность биотинидазы, аргининемия и множество других болезней. Из обязательного списка в такое исследование входит только фенилкетонурия.
Техника забора крови для расширенного скрининга ничем не отличается от порядка действий при плановом исследовании. Результаты анализа можно получить через 2–3 недели.
Расширенный неонатальный скрининг выявляет изменение концентрации метаболитов в ту или иную сторону, то есть повышенное или пониженное содержание этих веществ может говорить о наличии генетического заболевания. Например, при фенилкетонурии повышен фенилаланин.
Как и при обязательном скрининге, при серьезных отклонениях от нормы врач направляет малыша к узким специалистам для проведения дополнительных исследований и разработки схемы лечения, если диагноз подтвердится.
Генетический скрининг новорожденных особенно необходим, если раньше в семье были случаи наследственных заболеваний, пусть и в отдаленном прошлом. Довольно часто здоровые родители все же являются носителями дефектных генов и могут передать их потомству. Но даже если в вашей семье никто не страдал от генетических болезней, такой расширенный скрининг новорожденных сделать все равно стоит, поскольку риск наличия данных патологий у ребенка все равно есть.
Источник

Неонатальный скрининг, ласково именуемый в нашей стране «пяточка», является одним из первых важных исследований новорожденного. В России скрининг преимущественно направлен на выявление пяти наследственных болезней обмена: фенилкетонурии, врожденного гипотиреоза, врожденной дисфункции коры надпочечников (ВДКН), галактоземии и муковисцидоза. За рубежом этот список расширен до 50 различных заболеваний, в некоторых штатах Америки их свыше 60. Здоровый доношенный новорожденный допускается к скринингу на 4–5 сутки, недоношенный — на седьмой день после рождения. Заболевания, на выявление которых направлен скрининг, никак не проявляют себя в периоде новорожденности, но их ранняя диагностика и своевременно начатое патогенетическое лечение существенно влияют на прогноз и качество жизни ребенка. Помимо исследования крови проводится аудиометрия для оценки слуха и пульсоксиметрия для скрининга пороков сердца, но в данной статье мы преимущественно сосредоточимся на тестировании крови.
Разработка программы скрининга началась в шестидесятых годах прошлого века, когда Роберт Гатри создал технологию тестирования сухих отпечатков крови на фильтровальной бумаге. Первым заболеванием, которое стало кандидатом для массовой диагностики, была фенилкетонурия, так как ее раннее выявление и коррекция питания способны предотвратить развитие тяжелых неврологических нарушений. Затем к скринингу добавилось еще несколько заболеваний: врожденный гипотиреоз, ВДКН, галактоземия и муковисцидоз. Тандемная масс-спектрометрия (ТМС) позволила значительно расширить список заболеваний, добавив к болезням обмена веществ гемоглобинопатии, спинальную мышечную атрофию, тяжелый комбинированный иммунодефицит и др.
Таблица 1 | Рекомендуемая The American College of Obstetricians and Gynecologists (ACOG) скрининг-панель для врожденных заболеваний
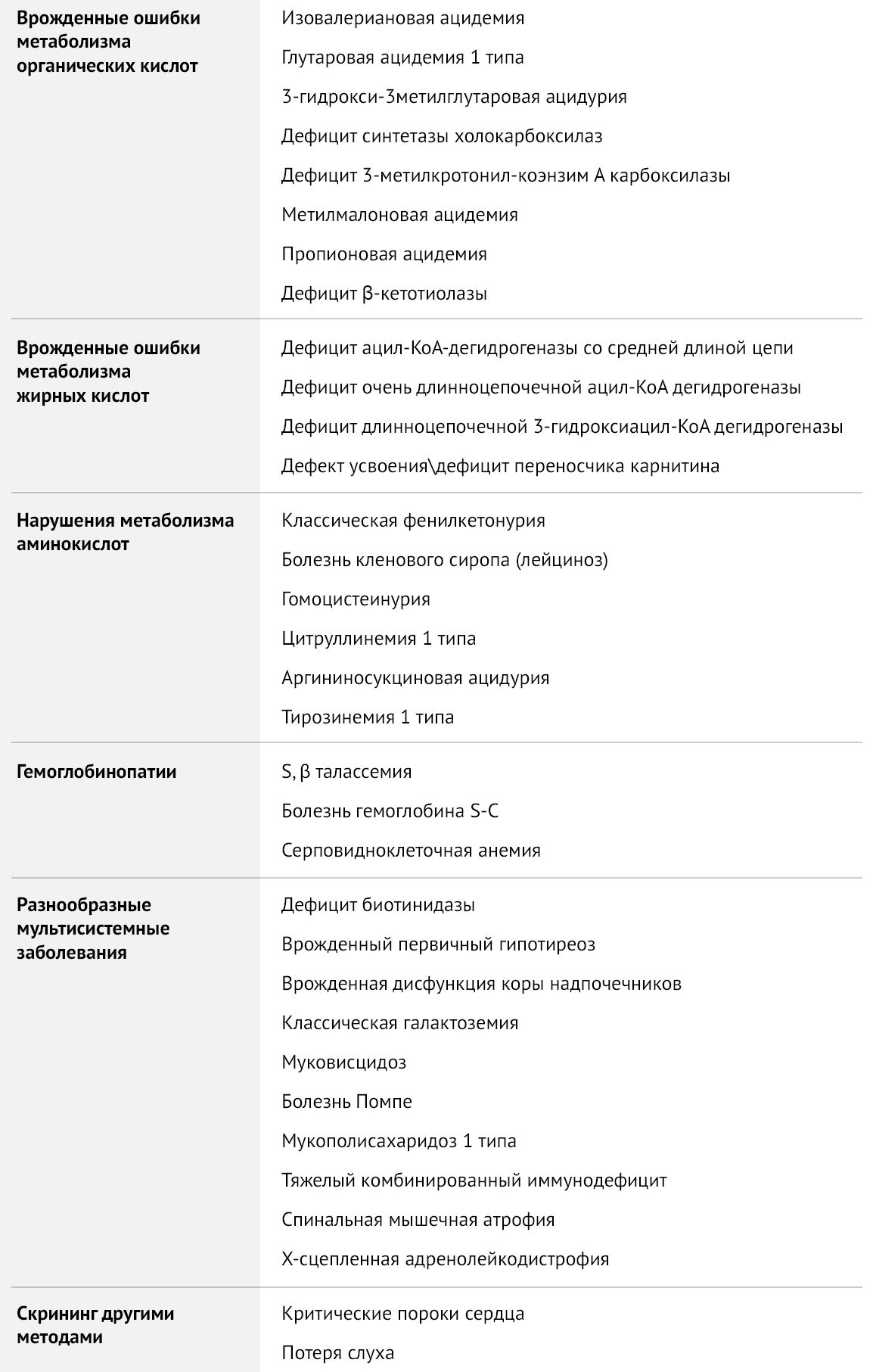
В данной статье будут рассмотрены два скрининга, доступные в нашей стране: обязательный, включающий тестирование на пять заболеваний (врожденный гипотиреоз, ВДКН, фенилкетонурия, галактоземия, муковисцидоз) и расширенный скрининг на наследственные нарушения метаболизма.
На 4–5 сутки после рождения здорового доношенного ребенка или на седьмые сутки жизни недоношенного ребенка проводится тестирование методом «сухого пятна».
Фенилкетонурия (в современной классификации ― ФАГ-зависимая ФКУ) обусловлена мутацией гена фенилаланингидроксилазы и относится к числу аминокислотных аминоацидопатий. В норме фенилаланин (ФА) путем реакций гидроксилирования превращается в тирозин, однако в случае мутации вышеназванного гена активность превращающего фермента снижается, создается дефицит тирозина одновременно с избытком ФА, образующего токсичные метаболиты (фенилацетат, фенилпируват, фениллактат). Снижение образования тирозина влечет за собой нарушение синтеза гормонов щитовидной железы, нейротрансмиттеров и пигментов меланоцитов, а избыток ФА приводит к дисбалансу аминокислот в тканях мозга, обусловленному торможением их всасывания в желудочно-кишечном тракте или нарушением реабсорбции из почечных канальцев, нарушению образования или стабилизации полирибосом, снижению синтеза миелина, норадреналина и серотонина. Также за счет конкурентного ингибирования создается дефицит тирозиназы, что в совокупности с дефицитом тирозина приводит к снижению образования меланина и гипопигментации.
Основной проблемой пациентов с ФКУ являются нарушения функции ЦНС: от сонливости, вялости, отсутствия аппетита в период манифестации в 2–6 месяцев до тяжелых нарушений психомоторного развития в будущем; нередко развиваются атаксия, гиперкинезы, тремор рук, парезы по центральному типу. Единственный способ предотвратить развитие вышеназванных нарушений — назначение гипофенилаланиновой диеты с момента рождения с поддержанием низкого уровня фенилаланина в течение всей жизни.
Рисунок 1 | Интерпретация результатов исследования на наличие фенилкетонурии
ВДКН обусловлена дефицитом ферментов и транспортных белков, участвующих в биосинтезе кортизола. Наиболее часто встречается дефицит 21-гидроксилазы, что в свою очередь приводит к дефициту кортизола и альдостерона и ответному увеличению секреции АКТГ и гиперплазии коры надпочечников. В условиях дефицита фермента происходит значительное накопление предшественников гормонов, что приводит к увеличению синтеза тестостерона, не зависящего от 21-гидроксилазы. В итоге у пациента формируется надпочечниковая недостаточность и гиперандрогения. Гормональным маркером дефицита 21-гидроксилазы является уровень 17-гидроксипрогестерона (17-ОНП), определяемый в рамках неонатального скрининга. Результат трактуется как положительный, если при двукратном тестировании образца уровень 17-ОНП у доношенных новорожденных составляет ≥ 20 нг/мл. У недоношенных детей при заборе крови на 7–8 сутки после рождения скрининговый результат трактуется как положительный при следующих уровнях 17-ОНП: на сроке 23–32 недели гестации ― ≥ 65 нг/мл; на сроке 33–36 недель гестации ― ≥ 40 нг/мл.
Врожденный гипотиреоз в большинстве случаев вызван дефектами самой щитовидной железы (первичный гипотиреоз). Причины первичного врожденного гипотиреоза можно в широком смысле классифицировать как неспособность щитовидной железы нормально развиваться (дисгенезия) или неспособность структурно нормальной щитовидной железы производить нормальные количества гормона (дисгормоногенез). Дисгенезия щитовидной железы, охватывающая весь спектр агенеза, гипоплазии и эктопии, является наиболее частой причиной врожденного гипотиреоза. В то время как это заболевание остается наиболее частой причиной врожденного гипотиреоза, частота возникновения дисгормоногенеза за последние несколько десятилетий увеличилась. В то время как на дисгормоногенез приходится только 15 % врожденного гипотиреоза, диагностированного в первые дни скрининга новорожденных, у 30–40 % младенцев, прошедших скрининг по современным протоколам, имеется эктопическая щитовидная железа, соответствующая одной из форм дисгормоногенеза. В отличие от дисгенезии щитовидной железы, при которой моногенная причина присутствует только у небольшого количества пациентов, дисгормоногенез часто возникает из-за генетического дефекта на каком-либо этапе синтеза тиреоидных гормонов.
Учитывая разнообразие функций тиреоидных гормонов в организме человека, врожденный гипотиреоз характеризуется разнообразием клинических проявлений с поражением всех органов и систем. При отсутствии своевременного лечения на первый план выходит задержка психомоторного и речевого развития, затем наступают отставание в физическом развитии и задержка полового развития. Основной задачей скрининга является наиболее раннее выявление детей с подозрением на врожденный гипотиреоз.

Рисунок 2 | Интерпретация результатов исследования на наличие врожденного гипотиреоза
Галактоземия — аутосомно-рецессивное наследственное нарушение обмена углеводов, при котором в организме накапливается избыток галактозы и ее метаболитов. В норме галактоза образуется в результате гидролиза лактозы в кишечнике либо в процессе ферментных реакций, обмена гликопротеинов и гликолипидов. Галактоза является материалом для образования клеточных мембран, нервной ткани, нервных окончаний и т. д. В результате ферментных реакций она превращается в глюкозу, и именно дефицит галактозо-1-фосфатуридилтрансферазы лежит в основе патогенеза данного заболевания. Метаболиты галактозы обладают повреждающим действием. Так, галактитол проникает в хрусталик глаза, приводя к повышению осмотического давления, электролитным нарушениям и денатурации белка с формированием катаракты. Другие метаболиты обладают гепато-, нейро- и нефротоксическим действиями, а также вызывают гемолиз эритроцитов. Тормозящее влияние метаболитов галактозы на углеводный обмен приводит к гипогликемии.

Рисунок 3 | Интерпретация результатов исследования на наличие галактоземии
Муковисцидоз — аутосомно-рецессивное заболевание, связанное с мутацией гена МВТР (трансмембранного регулятора муковисцидоза). МВТР является хлорным каналом, мутации гена которого нарушают не только транспорт, но и секрецию ионов хлора. При затруднении их прохождения через клеточную мембрану увеличивается реабсорбция натрия железистыми клетками, нарушается электрический потенциал просвета, что вызывает изменение электролитного состава и дегидратацию секрета желез внешней секреции. В результате выделяемый секрет становится чрезмерно густым и вязким. Поражаются все экзокринные железы организма: печень, поджелудочная железа, мочеполовая система, но наиболее ярко муковисцидоз проявляет себя со стороны органов дыхания, провоцируя бронхообструкцию, дыхательную и сердечную недостаточность, легочную гипертензию.

Рисунок 4 | Интерпретация результатов исследования на наличие муковисцидоза
Органические ацидемии — группа аутосомно-рецессивных наследственных заболеваний обмена, в основе патогенеза которых лежит дефицит ферментов, участвующих в метаболизме белков, что приводит к повышению уровня кетоновых тел, обладающих токсическим действием на различные органы и ткани, в частности, на ЦНС. Данные заболевания манифестируют уже в стадии декомпенсации, как правило, в период с первой недели до первого года жизни. Триггерами служат стресс, длительное голодание, инфекционные заболевания, иммунизация, реже — чрезмерное употребление белковой пищи. Проявляются преимущественно неврологической симптоматикой: нарушение сознания вплоть до комы, эпилептические приступы, нарушение мышечного тонуса, у детей старшего возраста — нарушения психоречевого развития, атаксия, очаговые неврологические симптомы, синдром Рейе (острая печеночная недостаточность, сочетающаяся с энцефалопатией), психические расстройства.
Нарушения окисления жирных кислот — врожденный дефект метаболизма из-за нарушения либо митохондриального β-окисления, либо транспорта жирных кислот с использованием карнитинового транспортного пути. Проявления зависят от нарушения метаболизма конкретной кислоты, но все они имеют общие черты и требуют схожей тактики лечения. В периоде новорожденности метаболические нарушения проявляются тяжелой кардиомиопатией, гипокетотической гипогликемией, дисфункцией печени в первые несколько дней или недель жизни, часто заканчиваясь летально. В младенческом и детском возрасте характерны эпизоды летаргии и рвоты, развивается дисфункция печени и гипокетотическая гипогликемия, энцефалопатия, что может привести к внезапной младенческой смерти. У подростков и во взрослом возрасте дебютируют эпизодическим рабдомиолизом, мышечной слабостью, миалгией. Лечение включает отказ от голодания, симптоматическую терапию развившихся осложнений и включение в рацион добавок, если это необходимо.
Болезнь кленового сиропа (она же лейциноз) — наследственное заболевание, обусловленное дефицитом дегидрогеназы кетокислот с разветвленной цепью и нарушением метаболизма лейцина, изолейцина, валина (аминокислоты с разветвленной цепью, АКЦР). Повышение уровня АКЦР и их метаболитов, в частности, кетокислот, приводит к кетоацидозу, атрофии ткани головного мозга, нарушению окислительного фосфорилирования в дыхательной цепи митохондрий. Избыток лейцина обладает нейротоксическим эффектом, вызывая дисфункцию астроцитов, апоптоз нейронов и блокируя транспорт через гематоэнцефалический барьер аминокислот, важных для синтеза нейротрансмиттеров.
Гомоцистеинурия — наследственное заболевание из группы аминоацидопатий, обусловленное нарушением метаболизма серосодержащих аминокислот, в частности, метионина. Дефицит цистатион-b-синтазы нарушает преобразование метионина в цистеин. Высокий уровень гомоцистеина связан с образованием некротически-дегенеративных участков в почках, селезенке, слизистой оболочке желудка и сосудах, активацией XII фактора свертывания, способствующего тромбообразованию.
Аргининосукциновая ацидурия вызывается мутациями в гене ASL, который кодирует фермент аргининосукцинатлиазу. Этот фермент катализирует превращение аргинино-янтарной кислоты в аргинин и фумарат на четвертом этапе цикла мочевины. Дефекты на этой стадии цикла мочевины приводят к накоплению в плазме аммиака, аргинино-янтарной кислоты, цитруллина и оротовой кислоты в моче, а также к дефициту аргинина в плазме. Ацидурия может иметь различную клиническую картину с началом в любом возрасте, включая период новорожденности. Состояние новорожденных обычно не вызывает подозрений в течение первых 24–48 часов после рождения, но в течение нескольких дней дебютирует тяжелая гипераммониемия, проявляющаяся летаргией, сонливостью, отказом от еды, рвотой, тахипноэ и респираторным алкалозом. Если не начать лечение, может произойти обострение летаргии, судороги, кома и смерть. Позднее начало ацидурии обычно индуцировано острой инфекцией, стрессом или высоким потреблением белка. Сообщалось также о поздних когнитивных дефектах или нарушениях обучаемости при отсутствии эпизодов гипераммониемии. У некоторых пациентов заболевание может протекать бессимптомно, несмотря на четкие биохимические признаки.
Тирозинемия 1 типа — заболевание, обусловленное дефицитом фумарилацетоацетатгидролазы, в результате чего происходит накопление высокотоксичных фумарил- и малеилацетоацетата, обладающих гепатотоксическим и канцерогенным действием. Конечные метаболиты — сукцинилацетон и сукцинилацетоацетат — являются митохондриальными токсинами, тормозящими фосфорилирование и блокирующими цикл Кребса. Накопление токсинов приводит к прогрессирующему заболеванию печени с развитием печеночной недостаточности, цирроза, тубулопатии с формированием ренальной тубулопатии, гипофосфатемического рахита, синдрома Фанкони. Острая тирозинемия сопровождается развитием гипертрофической кардиомиопатии. Кроме того, нарушается путь синтеза порфирина, ингибируется синтез порфобилиногена, что приводит к кризам, проявление которых напоминает порфирию. Все пациенты подвержены высокому риску развития гепатоцеллюлярной карциномы, вторичной по отношению к циррозу. Без своевременного лечения дети погибают в возрасте 10 лет.
- Rink B., Dukhovny S. Newborn Screening and the Role of the Obstetrician-Gynecologist //OBSTETRICS AND GYNECOLOGY. – 2019.
- Merritt J. L., II M. N., Kanungo S. Fatty acid oxidation disorders //Annals of translational medicine. – 2018.
- Еремина Е. Р. Клинический случай редкой органической ацидурии //Медицинская генетика. – 2018.
- Cherella C. E., Wassner A. J. Congenital hypothyroidism: insights into pathogenesis and treatment //International journal of pediatric endocrinology. – 2017.
- Чикулаева О. А. Федеральные клинические рекомендации по диагностике и лечению врожденного гипотиреоза у детей //Проблемы эндокринологии. – 2014.
- Клинические рекомендации “Фенилкетонурия и нарушения обмена тетрагидробиоптерина у детей”, 2017.
- Клинические рекомендации “Федеральные клинические рекомендации по оказанию медицинской помощи детям с галактоземией”, 2015
- Клинические рекомендации “Кистозный фиброз (муковисцидоз)”, 2020
- Клинические рекомендации “Гомоцистеинурия”, 2016
- Клинические рекомендации “Клинические рекомендации по ведению и терапии новорожденных с заболеваниями надпочечников”. 2016
- De Laet C. et al. Recommendations for the management of tyrosinaemia type 1 //Orphanet journal of rare diseases. – 2013.
- Nagamani S. C. S., Erez A., Lee B. Argininosuccinate lyase deficiency //Genetics in medicine. – 2019.
Нашли опечатку? Выделите фрагмент и нажмите Ctrl+Enter.
Источник