
Миоклонии у ребенка 2 года

Доброкачественная миоклоническая эпилепсия младенчества (ДМЭМ) – зависящая от возраста форма идиопатической эпилепсии, которая характеризуется генерализованными миоклоническими припадками. Этиология детально не изучена. Патология проявляется мышечными сокращениями верхних конечностей, шеи и головы длительностью в 1-3 сек. с частотою 2-3 раза в день. Общее состояние ребенка и его психофизическое развитие нарушается редко. Диагностика направлена на определение спайк- или полиспайк-волн на ЭЭГ. Основное лечение – медикаментозная монотерапия. Препараты выбора – вальпроаты, при их неэффективности используются бензодиазепины или производные сукцинимида.
Общие сведения
Доброкачественная миоклоническая эпилепсия младенчества (ДМЭМ) – редкая форма эпилепсии в педиатрии. Характерна только для определенной возрастной категории. Впервые заболевание выделено как отдельная нозологическая форма в 1981 году Дарве и Биором. Патология составляет менее 1% от всех форм эпилепсий и порядка 2% от ее идиопатических генерализованных форм. На данный момент в литературе описано около 100-130 случаев данного заболевания. ДМЭМ наблюдается у детей от 6 месяцев до 3 лет, в редких случаях возникает в возрасте до 5 лет. Представители мужского пола болеют в 1,5-2 раза чаще. Патология, как правило, хорошо поддается лечению и полностью купируется в старшем возрасте (в основном – после 6 лет). Осложнения в виде отставания в психомоторном развитии возникают редко и только при отсутствии терапии.

Доброкачественная миоклоническая эпилепсия младенчества
Причины ДМЭМ
Доброкачественная миоклоническая эпилепсия младенчества относится к числу генетически детерминированных заболеваний, передающихся по полигенному типу наследования. Является малоисследованной патологией, поскольку встречается довольно редко. ДМЭМ входит в группу идиопатических генерализованных эпилепсий, однако связи с другими нозологиями из этой группы не установлено. На данный момент неизвестно, мутация каких генов приводит к развитию ДМЭМ.
При сборе семейного анамнеза выясняется, что родители 40% больных страдают или страдали эпилепсией либо фебрильными припадками. Патогенетически развитие миоклонических атак обусловлено возникновением разряда быстрых генерализованных спайк-волн (СВ) или полиспайк-волн (ПСВ). Их частота составляет 3 Гц или более, а длительность – 1-3 сек. Волны формируются в лобных или теменных участках коры головного мозга. Сами атаки могут быть спонтанными или возникающими на фоне определенных (звуковых, тактильных или ритмичных световых) раздражителей.
Симптомы ДМЭМ
ДМЭМ диагностируется в возрасте от 6 месяцев до 3 лет. Развитие ребенка до появления первых миоклонических припадков проходит нормально. Примерно у 20% детей проявляются редкие судороги при рождении или в неонатальном периоде. Общее состояние пациента страдает редко, нарушения неврологического статуса не выявляются. Первые миоклонические атаки поражают верхние конечности, шею и голову, редко – ноги. Они могут иметь разную интенсивность, в том числе – у одного и того же ребенка во время разных эпизодов. Выраженность колеблется от еле заметных подергиваний до видимых фибрилляций.
Частота припадков составляет 2-3 раза в сутки с разными временными интервалами. Длинных серий атак не наблюдается. Возможна провокация атаки громким звуком, тактильной или ритмичной световой стимуляцией. После каждого эпизода может наблюдаться рефрактерный период длительностью от 20 до 120 сек. В этом временном промежутке даже интенсивная стимуляция не вызывает новый приступ. При этом часто наблюдается мышечная атония. Для заболевания характерно усиление миоклонических приступов при засыпании (дремоте) и их исчезновение в фазе медленного сна.
Выделяют рефлекторный и спонтанный варианты ДМЭМ. В первом случае миоклонические приступы развиваются после воздействия триггеров. Спонтанная форма возникает без каких-либо предикторов. На ранних этапах заболевания и при низкой интенсивности миоклоний родители и педиатр могут принимать атаки за нормальные моторные реакции ребенка. Относительно выраженные миоклонические приступы могут сопровождаться наклоном головы вперед, отводящим и приводящим движением, сгибанием рук, редко – плавным вращением глазных яблок. Часто родители отмечают характерное «кивание» головой продолжительностью от 1 до 3 сек., редко – до 10 сек. (у детей старшего возраста). В некоторых случаях единственным клиническим проявлением ДМЭМ становится длительное смыкание глаз.
При тяжелых формах возможна генерализация судорог, сопровождающаяся потерей равновесия, внезапным выпадением предметов из рук, редко – расстройствами сознания. В процесс иногда вовлекаются межреберные мышцы, передняя брюшная стенка и диафрагма, из-за чего нарушается дыхание и может выслушиваться экспираторный шум. Для ДМЭМ характерно увеличение интенсивности клинических проявлений до определенного возраста и их последующее полное исчезновение. При длительном течении заболевания возможно отставание в психомоторном развитии. Трансформация в другие формы приступов, в том числе в абсансы, не происходит даже на фоне отсутствия специфического лечения.
Диагностика ДМЭМ
Диагностика доброкачественной миоклонической эпилепсии младенчества заключается в сборе анамнестических данных и проведении инструментальных методов исследования. Физикальное обследование ребенка в интериктальный период неинформативно. Лабораторные тесты каких-либо отклонений от возрастной нормы не выявляют. Наибольшую диагностическую ценность имеет повторная полиграфическая видеоэлектроэнцефалография (видео-ЭЭГ), которая позволяет обнаружить спайк-волны и доказать наличие миоклонических приступов. При необходимости проводится провокационная проба с ритмической световой или тактильной стимуляцией.
Вне приступов (а изредка – во время них) данные ЭЭГ остаются в пределах нормы, спонтанные спайк-волны возникают редко. Во время медленного сна возможно усиление разрядов в коре головного мозга при сохранении их нормальной структуры, возникновение быстрых ритмов или формальных изменений. В быструю фазу (REM-сон) могут фиксироваться генерализованные разряды спайк-волн. С целью исключения органической патологии могут назначаться нейросонография, компьютерная и магнитно-резонансная томография. При наличии клинической симптоматики на протяжении длительного периода проводится оценка психомоторного развития.
Дифференциальная диагностика ДМЭМ осуществляется с криптогенными детскими судорогами, доброкачественным неэпилептическим миоклонусом, синдромом Леннокса-Гасто и миоклонически-астатической эпилепсией раннего детского возраста.
Лечение ДМЭМ
Лечение ДМЭМ, как правило, проводится в амбулаторных условиях, исключением являются частые и тяжелые миоклонические атаки, требующие постоянного наблюдения. Показана медикаментозная терапия при помощи противоэпилептических средств. Первую линию составляют препараты из группы вальпроатов (натрия вальпроат). Важную роль играет подержание стабильной концентрации действующего вещества в крови. Нерегулярное введение назначенных средств провоцирует новые приступы и формирование резистентности к дальнейшей терапии данным медикаментом. При недостаточной эффективности вальпроатов показаны препараты из группы бензодиазепинов (нитразепам) или производных сукцинимида (этосуксимид). Терапевтический курс предполагает лечение на протяжении 3-4 лет с момента возникновения первых припадков.
При выраженной чувствительности к ритмичным световым раздражителям длительность курса увеличивается. При минимальной активности приступов или их исключительно рефлекторном характере лечение может проводиться в сокращенные сроки или не назначаться вовсе. При рецидиве миоклонических атак в старшем возрасте после пройденного лечения рекомендован упрощенный вариант аналогичного терапевтического курса. Обязательным моментом является психологическая поддержка семьи, непосредственно влияющая на эффективность лечения и направленная на исключение триггеров для ребенка.
Прогноз и профилактика ДМЭМ
Специфической профилактики для доброкачественной миоклонической эпилепсии младенчества не разработано. Прогноз в большинстве случаев благоприятный, заболевание, как правило, заканчивается полным выздоровлением ребенка. Миоклонические атаки, возникающие на фоне звуковой или тактильной стимуляции, прогностически более благоприятны, чем спонтанные. Переход в другие формы эпилепсии нехарактерен. Острый период, при котором наблюдаются выраженные припадки, в среднем длится менее 12 месяцев. Более чем у 53% детей в возрасте 6 лет все симптомы ДМЭМ полностью исчезают. Примерно у 14% в дальнейшем наблюдаются задержка психического развития или нарушения в поведении, из-за чего пациенты вынуждены получать образование в специализированных учебных заведениях. Частота осложнений напрямую зависит от своевременности диагностики, эффективности проводимого лечения и психологического климата в семье, в первую очередь – взаимоотношений между ребенком и матерью.
Доброкачественная миоклоническая эпилепсия младенчества – лечение в Москве
Источник
Детская миоклоническая эпилепсия у детей. Синдром Леннокса-Гасто
Детская миоклоническая эпилепсия характеризуется короткими по продолжительности приступами, представляющими собой кратковременные, часто симметричные мышечные сокращения в сочетании с резким снижением тонуса мышц туловища и внезапным падением вперед, нередко служащим причиной травм лица и повреждения полости рта. Миоклоническая эпилепсия представляют собой гетерогенную группу заболеваний с различной этиологией и вариабельным прогнозом. Тем не менее можно выделить по крайней мере пять различных подгрупп, охватывающих широкий спектр миоклонической эпилепсии в детской популяции.
Доброкачественный миоклонус младенчества дебютирует в младенческом возрасте и проявляется в виде серий миоклонических приступов, вовлекающих мышцы шеи, туловища и конечностей. По внешним проявлениям миоклонус можно ошибочно принять за младенческие спазмы, однако на ЭЭГ у пациентов с доброкачественным миоклонусом младенчества нет отклонений от нормы. Прогноз благоприятный, психомоторное развитие детей соответствует возрасту, миоклония прекращается к 2-летнему возрасту. Антиэпилептическая терапия не показана.
Развитие детей до дебюта миоклонических приступов детского возраста соответствует возрастной норме. Течение беременности и родов у матери ребенка без особенностей. Раннее развитие без патологии. Средний возраст ребенка в дебюте приступов — около 2 лет, однако возможен дебют приступов у детей от 6 мес. до 4 лет. Частота миоклонических приступов широко варьирует: у одних пациентов приступы возникают ежедневно, несколько раз в день, у других детей отмечаются межприступные периоды продолжительностью до нескольких недель.
У небольшой части пациентов дебюту миоклонической эпилепсии предшествуют фебрильные судороги или генерализованные тонико-клонические афебрильные приступы. Примерно в 50 % случаев у детей с миоклоническими приступами периодически возникают тонико-клонические приступы. На ЭЭГ быстрые комплексы пик-волна с частотой 2,5 Гц или выше в сочетании с нормальной основной активностью фоновой записи в большинстве случаев. По крайней мере в 1/3 случаев имеется эпилепсия в семейном анамнезе, что позволяет предположить наследственную этиологию заболевания.
Долговременный прогноз относительно благоприятный. Умственная отсталость отмечается в небольшом проценте случаев, и более чем у 50 % пациентов через несколько лет наступает ремиссия. Однако у значительной части детей наблюдаются речевые нарушения, трудности с обучением, эмоциональные и поведенческие расстройства, что требует длительного катамнестического наблюдения и адаптации детей с привлечением специалистов различных направлений.
Сложная миоклоническая эпилепсия это гетерогенная группа заболеваний с плохим прогнозом. Дебют парциальных или генерализованных тонико-клонических приступов наблюдается на первом году жизни и в типичных случаях предшествует дебюту миоклонической эпилепсии. Генерализованные судороги часто возникают на фоне инфекционного заболевания с поражением верхних дыхательных путей и субфебрильной лихорадкой; часто отмечается эпилептический статус. Примерно в Уз случаев диагностируется задержка психического развития. В анамнезе детей часто встречается указание на гипоксически-ишемическую энцефалопатию в перинатальном периоде; при определении неврологического статуса характерны признаки поражения пирамидных путей, экстрапирамидные расстройства в сочетании с микроцефалией. Указание на эпилепсию в семейном анамнезе реже встречается в этой группе пациентов по сравнению с детьми с типичной миоклонической эпилепсией.
У некоторых детей выявляется комбинация частых миоклонических и тонических приступов, в этих случаях при обнаружении на ЭЭГ медленных комплексов пик-волна заболевание диагностируется как синдром Леннокса-Гасто. Этот синдром характеризуется триадой резистентных к терапии приступов, медленной пик-волновой активностью на ЭЭГ в периоде бодрствования и умственной отсталостью. У пациентов со сложной миоклонической эпилепсией на межприступной ЭЭГ выявляются медленные пик-волновые разряды и резистентность к антиэпилептической терапии.
Приступы отличаются стойким характером, частота умственной отсталости и поведенческих нарушений достигает практически 75 % среди всех пациентов. Назначение препаратов вальпроевой кислоты или бензодиазепиновых производных может привести к снижению частоты или уменьшению выраженности приступов. У пациентов с приступами, резистентными к антиэпилептической терапии, следует рассматривать вопрос о назначении кетогенной диеты.
– Также рекомендуем “Юношеская миоклоническая эпилепсия – синдром Янца. Болезнь Лафоры”
Оглавление темы “Эпилепсия у детей”:
- Первый и рецидивирующие приступы судорог у детей
- Парциальные приступы судорог у детей. Аура
- Роландическая эпилепсия у детей. Энцефалит Расмуссена
- Абсансы у детей. Генерализованные тонико-клонические приступы у ребенка
- Детская миоклоническая эпилепсия у детей. Синдром Леннокса-Гасто
- Юношеская миоклоническая эпилепсия – синдром Янца. Болезнь Лафоры
- Младенческие спазмы. Диагностика и прогноз
- Синдром Ландау-Клеффнера. Диагностика и лечение
- Механизмы развития эпилепсии у детей. Патогенез
- ЭЭГ в диагностике эпилепсии. Принципы лечения эпилепсии у детей
Источник
Здравствуйте!
Ребенок-мальчик 1г7мес.
Роды на 41 неделе по сроку. Раннее излитие вод. На третьей потуге у меня не было сил, стала неправильно тужится – в итоге ребенка выдавливали ( чуть чуть). Не кричал (сказали что это не критично). Апгар 7/8.
Вес 3720, рост 53 см.
Голову начал держать месяцев с 2х , видимо из за тонуса.
Сел сам в 6 мес.
7 мес встал, мало ползал.
В 11 мес пошел без поддержки.
Периодически ходит на носочках, сейчас есть небольшой вальгус обеих стоп.
Беременность : ОРВИ на сроках 3 нед, 26 нед, 32 нед с насморком и температурой 37,3. Лежала на сохранении : на сроках 5-9 недель – отслойка плаценты, на сроке 26 недель ( угроза выкидыша-тонус). Там же орвии заболела.
Инфекций ДО и вовремя беременности необнаружено. Вс анализы и обследования в норме.
На одних узи при беременности на сроке 38 нед видели небольшое маловодье, на других -норму.
С рождения и до 4-5 месяцев очень беспокойный. Списывали на колики.
С рождения установочная кривошея. Ушло полностью после курса массажа в 4 месяца. Тонус нижних конечностей.
УЗИ ГМ в возрасте 1мес, 3мес, 6 мес в норме.
Объем головы при рождении 35 см, на данный момент 1г7мес-48-49см.
С рождения мраморная кожа.
С рождения не мог пить воду -давился до 5 месяцев. Потом начал потихоньку пить. Со смесью такой проблемы не было.
Долго блендерили пищу, до года примерно, на куски – рвотный рефлекс и давился, не мог откашляться. Сейчас ест маленькие кусочки.
Появился рвотный рефлекс, если вытирать рот и лицо махровым полотенцем, потом любой тканью. Сейчас если при нем вытирать стол тряпкой-тоже рвотный рефлекс.
Если вытирать лицо или стол бумажной салфеткой или влажной детской салфеткой-то такого нет.
На данный момент ребенок развивается по норме, но очень эмоциональный, лабильный, повышенная тревожность. При осмотре двух неврологов- ставят астено -невротический синдром , понижен тонус в плечевой зоне, небольшой запас слов.
Хотя на своем языке или укроченными словами говорит много. Общителен, но пуглив. Энергичен. Небольшие проблемы с укладыванием-долго не уснуть, как будто ему не устроиться и не наити себе место. От груди отлуче в 1г6 мес. В 2 месяца перенес острый пиелонефрит, лечились в больнице
Долго одним делом не занимается, минут по 5-10. Все куда то бежит, лезет.
При недовольстве может завалиться на пол, или резко откинутся назад.
Пытаюсь отучить -не обращать внимание , либо отвлечь.
Очень ласковый, целует, обнимает родных, игрушки.
Любит петь и танцевать.
Беспокойство заключается вследующем:
Примерно недели 3 назад обратила внимание на небольшие мышечные сокращения после засыпания, которые периодически присутствуют всю ночь или весь дневной сон.
Сокращается то пальчик, то кисть, то палец ноги, то мышца на шее., то мышца от пчеча к локтю., то движение всеми пальчиками на ноге.
При этом дыхание ровное, ребенок не просыпается.
Один раз было как то много – по очереди, то пальчики на руке, то на ноге, то бровка, рот, глаз.
Возможно ли что это доброкачественные миоклонии? Нормально ли что все это длиться весь сон?
Нужно ли в обязательном порядке делать ЭЭГ?
На момент осмотра неврологов-этого либо не было, либо я не замечала.
Осмотр был в 1г6 мес.
Заранее благодарю за ответ.
Вот сейчас пишу- ребенок спит( дневной сон) – одновременно сократилась мышца от плеча к локтю и от бедра к колену. Не сильно, но заметно.Через минуту губа, опять рука((((
Очень беспокоюсь.
Источник
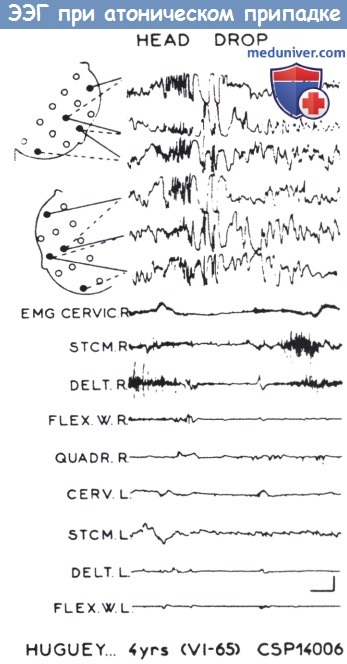
Эпилепсии с миоклоническими припадками у новорожденных, младенцевЭпилепсии, характеризующиеся главным образом истинными миоклоническими припадками, т.е. припадками, проявляющимися клинически очень кратковременными резкими мышечными сокращениями по типу шоковых, а на ЭЭГ — быстрыми комплексами спайк-волна или полиспайк-волна, начинаются в младенчестве или раннем детстве (Aicardi, 1996; Guerrini, Aicardi, 2003; Arzimanoglou et al., 2004). Такие эпилепсии часто путают с синдромом Леннокса-Гасто из-за часто повторяющихся коротких атак, которые могут быть причиной многократных падений, наличия разрядов типа спайк-волна при обоих состояниях и частой ассоциации с умственной отсталостью или ухудшением с припадками. Однако клинические и ЭЭГ-признаки миоклонических эпилепсий отличаются от проявлений СЛГ, и представление о контроле припадков и психическом развитии не всегда единообразны. Миоклонические припадки представляют собой внезапные молниеносные мышечные сокращения; могут вовлекаться мышцы всего тела (массивная миоклоническая атака) или только верхних конечностей, лица или век. Судороги обычно симметричны, но могут быть односторонними или локализованными в небольших группах мышц. На иктальной ЭЭГ выявляются пароксизмы полиспайк-волн. Иктальная электромиография демонстрирует очень короткие мышечные сокращения, за которыми следует период покоя в течение 100-350 мс. При относительной продолжительности этого периода расслабление мышц проявлется клинически, вызывая миоатонический припадок (Oguni et al., 1994, 1997). Иногда при так называемом «негативном миоклонусе» заметна только атоническая фаза (Guerrini и Aicardi, 2003). Клинически, атония различной интенсивности и длительности часто сопровождается миоклонической судорогой. Генерализованная и достаточно длительная атония приводит к падениям (дроп-атакам) и по этой причине отсутствует возможность отличить по клиническим признаками атонический припадок от миоклонического. Поэтому миоклоническую-астатическую эпилепсию можно считать вариантом миоклонической эпилепсии, при котором особенно выражен атонический компонент. Нозология миоклонических эпилепсий остается запутанной. Международная классификация выделяет три большие группы: тяжелые миоклонические эпилепсии или синдром Драве (Dravet et al., 1986, 1992а, 2005) (см. выше), доброкачественная миоклоническая эпилепсия (Dravet et al., 1992; Dravet, Bureau 2005), и эпилепсия с миоклоническими-астатическими припадками (Doose, 1992), к которым можно добавить неклассифицируемые случаи.
а) Доброкачественная миоклоническая эпилепсия младенцев. Доброкачественная миоклоническая эпилепсия (ДМЭ) характеризуется короткими миоклоническими припадками, спонтанными или вызванными шумом или контактом, начинающимися в возрасте от четырех месяцев до трех лет у детей с нормальным нервным развитием, преимуществен но у мальчиков (Dravet et al., 1992, 2005). Миоклонические припадки—единственный тип припадков при этом заболевании, за исключением редких простых фебрильных судорог у некоторых пациентов. Интериктальная ЭЭГ, включая регистрируемую во время сна, нормальна, спонтанные разряды спайк-волна встречаются редко. Миоклониям на ЭЭГ сопутствуют разряды в форме быстрых генерализованных спайк-волн или полиспайк-волн с частотой более 3 Гц, начинающиеся и заканчивающиеся вместе с миоклониями. Течение болезни доброкачественное, хорошо поддается монотерапии вальпроатом натрия, при необходимости в сочетании с этосуксимидом или бензодиазепинами. Лечение продолжается три или четыре года. Однако у отдельных детей сохраняются некоторые проблемы с учебой и легкая задержка развития. Фактически, далеко не всегда можно прогнозировать доброкачественное течение болезни, так как более чем у 10% больных сохраняются поведенческие и некоторые когнитивные нарушения (Dravet и Bureau, 2005). Неясно, должен ли термин «доброкачественная миоклоническая эпилепсия» ограничиваться случаями с исключительно миоклоническими припадками. Aicardi и Levy Gomes (1989) сообщали о 19 детях, преимущественно мальчиках, с высокой частотой припадков в предшествующих поколениях, у которых наблюдались нечастые тонико-клонические припадки и/или короткие абсансы вместе с частыми миоклоническими судорогами и относительно благоприятным исходом болезни. Эти случаи подтверждают существование спектра не вызванных какими-либо поражениями, вероятно, генетически детерминированных миоклонических эпилепсий. У некоторых младенцев припадки вызываются внезапными экстрацептивными или проприоцептивными стимулами. При данной «тактильной» или рефлекторной миоклонической эпилепсии (Ricci et al., 1995) прогноз благоприятный, и можно обойтись без лечения. Сходные с ДМЭ случаи могут возникать и у старших детей, и можно считать ДМЭ ранним проявлением идиопатической генерализованной эпилепсии (Arzimanoglou et al., 2004), эквивалентом ювенильной миоклонической эпилепсии, как предложили Dravet и Bureau (2005). Генетические причины доброкачественной миоклонической эпилепсии (ДМЭ) неизвестны. Случаи заболевания редки, и не описано ни одного случая семейной заболеваемости. Arzimanoglou et al. (1996) описали семью, в которой у пробанда была эпилепсия с миоклоническими-астатическими припадками, а его младший брат перенес типичную ДМЭ с благоприятным исходом.
б) Миоклоническая-астатическая эпилепсия. Миоклоническую-астатическую эпилепсию (возможно, более точно эпилепсию с миоклоническими-астатическими припадками), вероятно, лучше отнести к категории генерализованных, не связанных со структурными поражениями миоклонических эпилепсий, а не к отдельным синдромам (Guerrini и Aicardi, 2003; Arzimanoglou et al., 2004; Guerrini et al., 2005). Термин «миоклоническая-астатическая» впервые использовали Doose et al. (1970) в публикации о «центроцефальном миоклоническом-астатическом petit mal» и позже развили Doose и Baier (1987) в статье «Генетические факторы эпилепсий с первивично генерализованными малыми припадками». Первоначально так определяли форму наследственной, не связанной с повреждениями генерализованной эпилепии, и, возможно, включали случаи, которые сейчас называют тяжелыми или доброкачественными миоклоническими эпилепсиями, так же как и случаи, соответствующие современной концепции миоклонической-астатической эпилепсии (Guerrini и Aicardi, 2003). Заболевание начинается позже, чем ДМЭ или синдром Драве, обычно в возрасте от одного года до пяти лет, чаще у мальчиков. Оно характеризуется клинически преобладанием чисто миоклонических или/и миоастатических припадков; припадки могут вызывать падения или, если припадок кратковременный, серию эпизодов свисания головы оседания на колени. Часто этим типам припадков сопутствуют другие, включая генерализованные тонико-клонические атаки, атипичные абсансы и эпизоды неконвульсивного эпилептического статуса. Тонические припадки не являются преобладающими, но в некоторых сериях наблюдаются относительно часто (Kaminska et al., 1999; Oguni et al., 2001a). Позднее авторы отметили, что они возникали максимум в двух третях их случаев. Астатические припадки (дроп-атаки) могут иметь различные механизмы (астатические, миоклонические или тонические), но их невозможно определить без полиграфической записи. Тонические припадки могут более часто наблюдаться у детей с неблагоприятным исходом, но они также наблюдаются почти у 30% детей с благоприятным исходом заболевания (Kaminska et al., 1999). На ЭЭГ выявляются быстрые (<3 Гц) генерализованные кратковременные (обычно меньше 4-5 секунд) разряды спайк-волна или полиспайк-волна. Doose подчеркивает наличие бипариетальных тета-ритмов. Возможна ремиссия почти в 60% случаев в течение нескольких лет с нормальными когнитивными функциями (Oguni et al., 2001а; Guerrini et al., 2005). Однако в начале заболевания могут возникнуть трудности контроля падений. Они могут стать причиной травм и являются серьезной практической проблемой. У некоторых пациентов заболевание протекает тяжело, с частыми припадками и нарушениями обучения (20%) или умственной отсталостью (20%), но в начале заболевания исход предсказать невозможно. Эффективность антиэпилептических препаратов вариабельна. Препаратом первого выбора считается вальпроат натрия или, если необходимо, комбинация вальпроата натрия и ламотриджина. При миоклони-ческих припадках и абсансах доказана эффективность этосуксимида и бензодиазепинов, особенно клобазама. Наконец, интересной альтернативной могут быть топи-рамин и леветирацетам. Следует избегать назначения карбамазепина, вигабатрина, окскарбазепина и габа-пен гина (Arzimanoglou et al., 2004; Guerrini et al., 2005). в) Неклассифицированные формы. Ряд случаев миоклонических эпилепсий у младенцев и в раннем детском возрасте остается неклассифицированными, также существуют промежуточные между разными синдромами формы (Arzimanoglou et al., 2004). Их нужно отделять от других синдромов с частыми короткими атаками и повторяющимися падениями, особенно синдрома Леннокса-Гасто. Термин «миоклоническая-астатическая эпилепсия» все еще не имеет четких клинических или ЭЭГ признаков, он произвольно и с трудом дифференцируется от других миоклонических форм и может быть применен ко многим миоклоническим эпилепсиям. Дегенеративные миоклонические эпилепсии с прогрессивным течением обсуждаются в отдельных статьях на сайте. – Вернуться в оглавление раздела “Неврология.” Редактор: Искандер Милевски. Дата публикации: 3.1.2019 Оглавление темы “Эпилепсия и эпилептические припадки у детей.”:
|

Источник